थैलेसीमिया की परिभाषा
थैलेसीमिया एक आनुवंशिक रूप से संचरित रक्त विकार है जिसमें शरीर हीमोग्लोबिन के असामान्य रूप का संश्लेषण करता है।
जैसा कि ज्यादातर लोग जानते हैं, हीमोग्लोबिन लाल रक्त कोशिकाओं में निहित एक प्रोटीन है, जो रक्त में ऑक्सीजन के परिवहन के लिए आवश्यक है। थैलेसीमिया से पीड़ित विषयों में, हीमोग्लोबिन का उत्परिवर्तित रूप एनीमिया तक लाल रक्त कोशिकाओं के क्रमिक लेकिन कठोर विनाश का कारण बनता है।
चिकित्सा आँकड़ों से यह स्पष्ट है कि थैलेसीमिया मुख्य रूप से मध्य पूर्वी देशों, अफ्रीकी देशों के निवासियों और उन सभी लोगों को प्रभावित करता है जो दलदली जगहों को आबाद करते हैं (आश्चर्य की बात नहीं, थैलेसीमिया भी कहा जाता है) मेडिटेरेनियन एनीमिया).
वर्गीकरण और कारण
दोषपूर्ण प्रोटीन सबयूनिट (जो हीमोग्लोबिन बनाता है) के अनुसार, थैलेसीमिया के दो रूपों को प्रतिष्ठित किया जाता है; विश्लेषण के साथ आगे बढ़ने से पहले, आइए कुछ बहुत महत्वपूर्ण अवधारणाओं को स्पष्ट करने के लिए एक कदम पीछे हटें।
हीमोग्लोबिन उत्कृष्ट वाहक है, जिसका उपयोग रक्त में ऑक्सीजन के परिवहन के लिए किया जाता है; यह दो प्रोटीनों से बना होता है, जिन्हें अल्फा-ग्लोबुलिन और बीटा-ग्लोब्युलिन के रूप में जाना जाता है।
थैलेसीमिया तब होता है जब इन प्रोटीनों में से एक या दोनों के उत्पादन को नियंत्रित करने वाले एक या अधिक जीन दोषपूर्ण (उत्परिवर्तित) होते हैं।
थैलेसीमिया प्रोटीन के डीएनए उत्परिवर्तन के कारण होता है जो हीमोग्लोबिन बनाते हैं: इन परिवर्तनों का हीमोग्लोबिन के शारीरिक संश्लेषण पर भारी प्रभाव पड़ता है और, एरिथ्रोसाइट्स को नष्ट करके, एनीमिया का कारण बनता है।
थैलेसीमिया का वर्गीकरण दो महत्वपूर्ण कारकों के आधार पर किया जाना चाहिए:
- माता-पिता से विरासत में मिले उत्परिवर्तित जीनों की संख्या
- शामिल प्रोटीन का प्रकार (अल्फा या बीटा हीमोग्लोबिन)
अल्फा थैलेसीमिया
थैलेसीमिया के "अल्फा" रूप में - जिसमें हीमोग्लोबिन के 4 "अल्फा" गोलाकार सबयूनिट (गुणसूत्र 16 पर) उत्परिवर्तित हो सकते हैं - एक या अधिक दोषपूर्ण जीन शामिल होते हैं; प्रत्येक गोलाकार सबयूनिट एक जीन से स्पष्ट रूप से एन्कोडेड होता है, इसलिए जिन जीनों को शामिल किया जा सकता है वे 4 हैं।

जब तीन या चार जीन शामिल होते हैं तो सामान्य लक्षण तस्वीर और अधिक गंभीर हो जाती है: पहले मामले में, हम बात करते हैं "हीमोग्लोबिन एच रोग"(मध्यम या गंभीर लक्षणों के साथ)। जब सभी चार जीन शामिल होते हैं, तो रोग कहलाता है अल्फा थैलेसीमिया मेजर: इसी तरह की स्थितियों में, नवजात शिशु जन्म से कुछ समय पहले या उसके तुरंत बाद मर जाता है।
बीटा थैलेसीमिया
थैलेसीमिया का बीटा रूप, जैसा कि अनुमान लगाया जा सकता है, तब होता है जब बीटा श्रृंखलाओं की संरचना में शामिल जीन उत्परिवर्तित होते हैं (गुणसूत्र 11 के स्तर पर): इस मामले में, केवल दो जीन प्रभावित हो सकते हैं। यदि केवल एक जीन को बदला जाता है, तो इसे कहा जाता है बीटा-थैलेसीमिया माइनर, जिसमें रोगी कोई प्रासंगिक लक्षण नहीं होने की शिकायत करता है। इसी तरह अल्फा संस्करण के लिए, हीमोग्लोबिन की बीटा श्रृंखला बनाने वाले दोनों जीनों की भागीदारी के परिणामस्वरूप एक बीटा-थैलेसीमिया मेजर (या कूली की रक्ताल्पता), जो गंभीर और गंभीर लक्षणों को दर्शाता है; इस मामले में, हालांकि, लक्षण आमतौर पर जन्म के कुछ वर्षों के बाद शुरू होते हैं।
वह वीडियो देखें
- यूट्यूब पर वीडियो देखें
लक्षण
अधिक जानकारी के लिए: थैलेसीमिया के लक्षण
थैलेसीमिया एक बहुत ही गंभीर वंशानुगत बीमारी है, इतना अधिक कि इसके कुछ प्रकार, जैसे अल्फा-थैलेसीमिया मेजर, प्रसव के दौरान या जन्म के तुरंत बाद बच्चे की मृत्यु का कारण बन सकते हैं। हालांकि, बीटा-थैलेसीमिया मेजर वाले शिशु जीवित रह सकते हैं और विकसित हो सकते हैं। जन्म के कुछ वर्षों के भीतर पहले लक्षण (गंभीर एनीमिया)।
यदि केवल एक जीन को बदल दिया जाता है, दोनों अल्फा और बीटा थैलेसीमिया के रूप में, रोगियों को किसी भी महत्वपूर्ण लक्षण की शिकायत नहीं होती है; केवल रोगी से लिए गए रक्त के नमूने के सूक्ष्मदर्शी विश्लेषण के माध्यम से, एरिथ्रोसाइट्स के आकार और संरचना में असामान्यता होती है, जो सामान्य से बहुत छोटी होती है।
एनीमिया के अलावा, थैलेसीमिया के रोगियों को निम्नलिखित लक्षणों में से एक या अधिक का अनुभव हो सकता है: थकान, मनोदशा में बदलाव (चिड़चिड़ापन), विकास की विफलता, चेहरे की हड्डी की विकृति, पीलिया, सांस की तकलीफ और गहरे रंग का मूत्र।
गंभीरता के मामलों में, थैलेसीमिया से पीड़ित रोगी की रोगसूचक तस्वीर खराब हो सकती है, वास्तविक हड्डी विकृति पैदा करने के बिंदु तक, विशेष रूप से चेहरे और खोपड़ी पर; थैलेसीमिया "अस्थि मज्जा के असामान्य विस्तार को बढ़ावा दे सकता है, दोनों अस्थि द्रव्यमान को नाजुक बनाकर और अस्थि भंग के जोखिम को अत्यधिक बढ़ाकर।
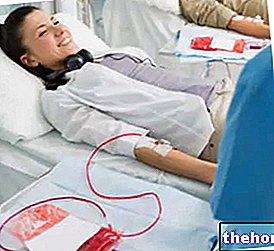
थैलेसीमिया की जटिलताओं के बीच, लोहे के संभावित संचय (हेमोक्रोमैटोसिस), एक अभिव्यक्ति दोनों ही रोग और रोगी को आवर्ती रक्त आधान की आवश्यकता होती है, का भी उल्लेख किया जाना चाहिए।
थैलेसीमिया अक्सर स्प्लेनोमेगाली का कारण बनता है, यानी प्लीहा में एक अतिरंजित वॉल्यूमेट्रिक वृद्धि: अक्सर, इस रोग संबंधी नैदानिक स्थिति में स्प्लेनेक्टोमी की आवश्यकता होती है, अंग का शल्य चिकित्सा हटाने। जैसा कि हम जानते हैं, प्लीहा रक्त कोशिकाओं के संश्लेषण के लिए उपयोग किया जाने वाला एक महत्वपूर्ण अंग है। और एंटीबॉडी, संक्रमण नियंत्रण के अलावा: इसका निष्कासन, स्पष्ट रूप से, बैक्टीरिया और वायरल अपमान के खिलाफ रक्षा कार्य में कमी का पक्षधर है, जिससे विषय संक्रमण के प्रति अधिक संवेदनशील हो जाता है। हालांकि, यह ध्यान दिया जाना चाहिए कि थैलेसीमिया भी जोखिम को बढ़ाता है संक्रमण के अनुबंध के लिए: थैलेसीमिया के संदर्भ में तिल्ली के छांटने की स्थिति में, संक्रमण की संभावना अत्यधिक बढ़ जाती है।

निदान
यदि पिता और/या माता थैलेसीमिया से प्रभावित हैं, तो संतान को रोग के संचरण की संभावना बहुत अधिक होती है। हमने विश्लेषण किया है कि थैलेसीमिया के सभी रूप जन्म से ही एक सटीक रोगसूचकता के साथ शुरू नहीं होते हैं: इसी तरह की स्थितियों में, संदिग्ध थैलेसीमिया के मामले में, रोगी को विशिष्ट परीक्षणों और परीक्षाओं की एक श्रृंखला के अधीन करना संभव है, जिसका उद्देश्य नैदानिक मूल्यांकन ( जैसे हीमोग्लोबिन A2 का निर्धारण, जो बीटा-थैलेसेमिक जीन ले जाने वाले स्वस्थ विषयों में ऊंचा होता है)।
शारीरिक परीक्षाओं में, प्लीहा का चिकित्सकीय तालमेल कभी-कभी थैलेसीमिया का पता लगा सकता है: स्प्लेनोमेगाली, जैसा कि पहले उल्लेख किया गया है, भूमध्यसागरीय एनीमिया के लिए पहला अलार्म संकेत है। रक्त परीक्षण अधिक विशिष्ट और सटीक होते हैं: थैलेसीमिया से रक्त के नमूने में, लाल रक्त कोशिकाएं, जब एक माइक्रोस्कोप के नीचे देखी जाती हैं, छोटी दिखाई देती हैं और उनका आकार असामान्य होता है। इसके अलावा, थैलेसीमिया से पीड़ित रोगी की सावधानीपूर्वक रक्त गणना से गंभीर एनीमिया का पता चलता है: यह परीक्षण रक्त में आयरन की मात्रा के लिए, रोग के नैदानिक मूल्यांकन के लिए डीएनए विश्लेषण करने और "हीमोग्लोबिन" के संभावित उत्परिवर्तन का मूल्यांकन करने के लिए उपयोगी है। .
दूसरी ओर, हीमोग्लोबिन के वैद्युतकणसंचलन से ऑक्सीजन ले जाने वाले प्रोटीन के असामान्य आकार का पता चलता है।
थैलेसीमिया के कुछ प्रकारों का वैद्युतकणसंचलन के साथ निदान नहीं किया जा सकता है: इस मामले में, रोगी को "म्यूटेशनल विश्लेषण" परीक्षण के अधीन किया जाएगा, जो थैलेसीमिया का पता लगाने और पता लगाने के लिए उपयोगी है।
दवाएं और उपचार
यह भी देखें: थैलेसीमिया के इलाज के लिए दवाएं
आनुवंशिक रूप से संचरित रोग होने के कारण, यह समझ में आता है कि - फिलहाल - ऐसी कोई दवा नहीं है जो रोग को उलट सके; हालांकि, रोगी के जीवन की गुणवत्ता में सुधार करते हुए, लक्षणों को नियंत्रित करना संभव है। दूसरे के बजाय एक उपचार का चुनाव थैलेसीमिया के प्रकार और लक्षणों की गंभीरता पर निर्भर करता है।
थैलेसीमिया के हल्के रूप में (जिसमें, उदाहरण के लिए, केवल एक जीन बदला जाता है) किसी दवा की आवश्यकता नहीं होती है, क्योंकि रोगी लक्षणों की शिकायत नहीं करता है। ऐसी परिस्थितियों में, अभी भी नियमित रूप से आवश्यक जांच करने की सलाह दी जाती है; कभी-कभी, कभी-कभी रक्त आधान कभी-कभी उपयोगी होता है (विशेषकर शल्य चिकित्सा और प्रसव के मामले में)।
मध्यम या गंभीर रोगसूचक रूपों के लिए, उपचार का तरीका अलग है, और इसके लिए बार-बार रक्त आधान या, गंभीर मामलों में, स्टेम सेल प्रत्यारोपण की आवश्यकता हो सकती है।
- रक्त आधान: यह चिकित्सीय दृष्टिकोण गंभीर जटिलताएं भी पैदा कर सकता है, क्योंकि बार-बार आधान रक्त में लोहे के एक रोग संचय (हेमोक्रोमैटोसिस) का पक्ष ले सकता है, जिसके लिए लोहे के भंडारण को समाप्त करने के उद्देश्य से विशिष्ट उपचार की आवश्यकता होती है, जिसे थेरेपी केलेटर के रूप में जाना जाता है। डेफेरिप्रोन)। अधिक जानकारी के लिए: हेमोक्रोमैटोसिस के उपचार के लिए दवाओं पर लेख पढ़ें।
- अस्थि मज्जा प्रत्यारोपण: सबसे गंभीर मामलों के लिए आरक्षित, जिसमें थैलेसीमिया शरीर में गंभीर शिथिलता पैदा करता है।
-cosa-significa-quando-preoccuparsi.jpg)